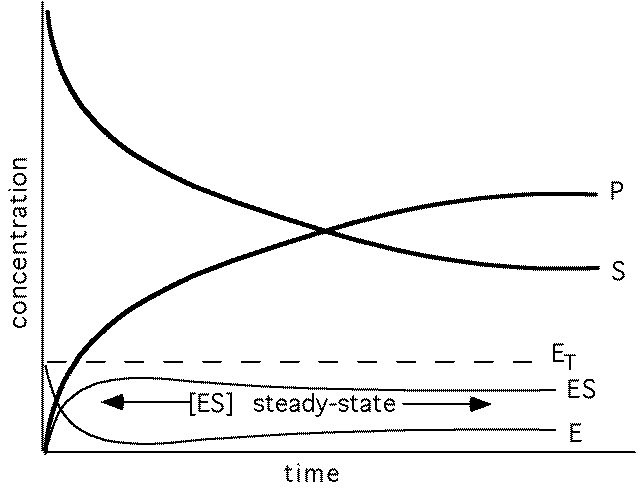
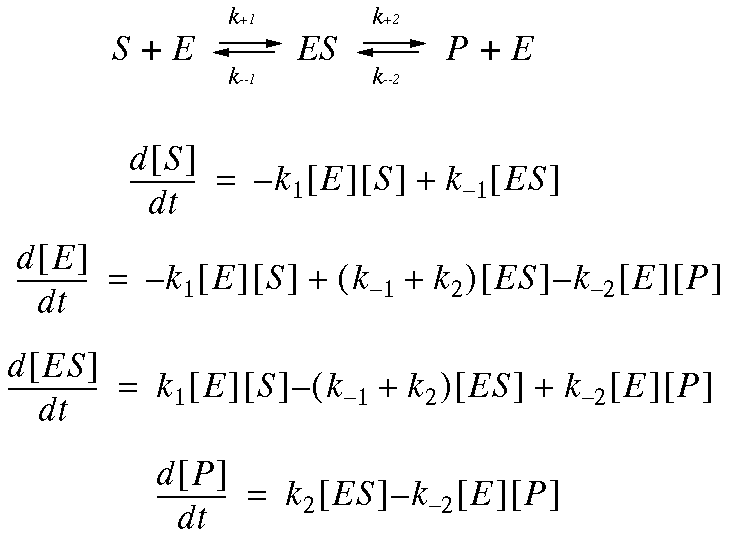Kinetic Modeling: Definitions
From a modeling perspective, biochemical networks are a set of
chemical species that can be converted into each other through
chemical reactions. The focus of biochemical network models is
usually on the levels of the chemical species and this usually
requires explicit mathematical expressions for the velocity at which
the reactions proceed.
The most popular representation for these models uses ordinary
differential equations (ODEs) to describe the change in the
concentrations of the chemical species.
Each chemical species in the network is represented by an ODE that
describes the rate of change of that species along time. The ODE is
composed by an algebraic sum of terms that represent the rates of
the reactions that affect the chemical species. For a chemical
species X :

where si
is a stoichiometry coefficient that is the number of molecules of
X consumed or produced in one cycle of reaction i,
with a positive sign if it is produced or negative if consumed, and
vi is the velocity of reaction i
. Obviously, for reactions that do not produce or consume X
the corresponding si
is zero.
The velocity of each reaction is described by a rate law that
depends on the concentrations of the reaction substrates, products,
and modifiers.
These ODE models can be used to simulate the dynamics of the
concentrations of the chemical species along time given their
initial values.
Kinetic
simulation of biochemical systems involves the following basic
steps:
-
Identification of the kinetic problem: This step involves the
determination and output variables as well as the intermediates. Key
species and physical properties to be simulated are determined and
tabulated.
-
Model formulation: Although that does not have to be the case, most
studies employ the reaction rate equations (RRE) to model
biochemical systems. In the RRE, one simply defines the changes in
the concentrations (or equivalently the number of molecules) as a
function of time and location. We note that constraints can be
embedded in RRE using a Lagrange undetermined multipliers type
formalism.
-
Choosing a method: At this step a decision needs to be made as to
whether to adopt a deterministic or stochastic formulation.
Although it is still relatively uncommon, a hybrid approach
which moves between deterministic and stochastic regimes is
another possibility. In the deterministic approach, a reasonable
time step is chosen adaptively, based on the estimated local
error according to a differential equation model. In contrast,
discrete stochastic simulations model each individual reaction
event. Using the probabilities of the reactions (called the reaction propensities),
one determines the occurrence statistics of the involved reactions.
This is generally done in one of two ways: In next reaction type
methods, the sequence of the reactions are chosen according to the
reaction propensities and the reaction times are computed to of the
elapsed time step and compute how many times the involved reactions
occur according to their probabilities.
-
Simulation: Integrating the RRE deterministically involves choosing
an appropriate algorithm from a wide selection of sophisticated
numerical methods for systems of ODEs and DAEs. Highly efficient and
reliable software is readily available, although one must usually
determine whether or not the problem is stiff and then choose the
software accordingly. Roughly speaking, an ODE or DAE system is
stiff if it involves a wide range of time scales and the fastest of
those scales correspond to stable processes. In chemically reacting
systems, the wide range of time scales can come from some reaction
rates being several orders of magnitude or more greater than others.
A solver which is designed for non-stiff systems will run very
slowly (because it needs to choose time steps on the scale of the
fastest process in the system to maintain stability for its explicit
formulas), if it is applied to a stiff problem. The cure for
stiffness is to approximate the differential equation using implicit
methods. The software that is available for the accurate solution of
stiff ODE and DAE systems always makes use of implicit methods.
Stochastic simulation algorithms are not as advanced as the
deterministic integrators and they do not scale well with the model
size and complexity. Even though recent algorithmic improvements
have led to impressive gains, stochastic simulations are still too
slow for most realistic systems. However, as discussed in the
previous section, fluctuations can be large in biological systems
and, more importantly, stochastic variations may have implications
for biological functions and responses. Therefore, when feasible,
stochastic simulation methods should be preferred. This is
particularly true for processes that involve small number of
regulatory molecules with large variations in their expression
levels.
-
Trajectory analysis: Time courses
obtained during the simulations are catalogued and combined to
derive the statistical distributions of the desired
output/prediction quantities, such as the concentrations of
experimentally monitored species and the resulting material/mass
flow in the system or the probability distribution of the reactions.
Similarly, from simulation results for different model parameter
sets, one can study the sensitivity to parameter variations.
Note:
Texts and figures in this page have been chosen
from the following references:
Kinetic Modeling of Biological Systems (Methods
Mol Biol. 2009, 541, 311–335)
|
Do you want reading more?
There are some comprehensive references which
could be helpful in more understanding kinetic modeling methods.
We recommend reading the following
references for better understanding: |